
 by: Parent Project Muscular Dystrophy
by: Parent Project Muscular Dystrophy
During PPMD’s Annual Advocacy Conference we had the opportunity to host a Gene Therapy Forum with expert clinicians, researchers, industry stakeholders, and government representatives taking part in the discussion.
This meeting continues PPMD’s in-depth look at this promising technology as part of our ongoing Gene Therapy Initiative. Below is a recap of the day-long forum.
Later this year, PPMD will host a series of community updates so that we can continue to follow the progress of this technology.
The Regulatory Landscape

Celia Witten, MD, PhD, Deputy Director at FDA within the Center for Biologics Evaluation and Research (CBER), provided an overview of the regulatory landscape around gene therapy (download presentation). Work within gene therapy has rapidly grown over the past few years, with CBER receiving over 200 new gene therapy product submissions in 2018, nearly doubling the 2017 submissions. There have also been three approved gene therapy treatments: Kymriah, Yescarta, and Luxturna.
With the influx of submissions has come a greater demand on CEBR regarding Gene Therapy, to that end CBER has been addressing these pressures through various pathways, such as:
- The July 2018 draft guidance on gene therapy (six guidance documents, one focused on rare disease, such as Duchenne)
- Facilitating numerous workshops to try and address potential issues
- Developing working relationships with international regulators to share information, best practices and promote regulatory convergence
Dr. Witten also provided an overview of the areas in the 21st Century Cures Act that pertain to gene therapy, particularly the ‘Advancing New Drug Therapies and Regenerative Medicine Advanced Therapy’ clauses. These areas provide information on expedited approval programs, which the FDA recently released further guidance on these issues to enhance understanding. CBER is also invested in improving manufacturing processes for drug availability. This legislation, along with CBER’s efforts, highlights the continued support of the FDA to try and bring safe and effective therapies to those in need as soon as possible.
Micro-dystrophin gene therapy in Duchenne
The audience also listened to updates regarding all three current micro-dystrophin gene therapy trials underway (download overview). At this stage of development, safety remains the primary outcome for all three trials. All the companies stressed their investments being made into manufacturing to ensure product availability should the products come to market. Although unique to each study, there are three main components to these drugs:
- Vector: The viral capsid being utilized to transport the transgene into the muscle cells. Both Pfizer and Solid are using AAV9, whereas Sarepta is using AAVrh74.
- Transgene: Each study is delivering a unique version of a truncated microdystrophin gene, which will result in different versions of truncated dystrophin protein being expressed. The efficacy differences between these transgenes remains to be seen.
- Promoter: A DNA sequence that controls where the microdystrophin gene is being expressed. Although each company is using a unique promoter, the region of dystrophin expression is intended to be restricted to the muscle.
Three Current Micro-dystrophin Gene Therapy Trials:
- Pfizer – Have completed dosing of six patients and will be sharing the initial data in June 2019. If the safety and expression outcomes of the current 1b study are met, Pfizer will aim for a Phase 3 global confirmatory study in 2020.
- Sarepta Therapeutics – Preliminary data from the initial four patients dosed at Nationwide Children’s Hospital has shown a favorable safety profile at one year. The preliminary data shared will require further scrutiny to confirm results, but dystrophin-positive fibers, protein expression, CK levels, and timed-function test scores all look positive in early stages. A second 24-patient placebo-controlled study is underway with nine subjects currently enrolled.
- Solid Biosciences – Enrolled six patients (three on active treatment, three on delayed control), including non-ambulatory subjects. Preliminary analysis of three-month biopsies detected low levels of microdystrophin in treated patients. Currently working on dose escalation that will be delivered to Cohort 2 and the delayed control treatment groups.
A Closer Look at AAV Delivery
Dr. Lee Sweeney, PhD, University of Florida, Chair of PPMD’s Scientific Advisory Board, shared AAV insights from pre-clinical models. Dr. Sweeney discussed other molecules that can be delivered via AAV. CRISPR/Cas9 can be utilized to correct nonsense and other small mutations. U7 snRNA can mediate exon skipping through a similar ‘one-time’ delivery as current microdystrophin strategies. Although micro-dystrophin has the potential to treat a large population of those with Duchenne, a number of individuals may gain more benefit from corrections via CRISPR or exon skipping dependent on their mutations and the dystrophin protein produced. Of course, with all of these AAV delivered drugs there are still significant hurdles.
- In older patients, the delivery of AAV can be impeded by fibrosis in the muscle.
- Neutralizing anti-bodies to AAV can impact the ability to receive gene therapy and preclude re-dosing.
- It is currently unknown how long the treatment will last, even when comparing to pre-clinical models, so redosing will likely be necessary.
Both Tim Cripe, MD, PhD, Nationwide Children’s Hospital (download presentation), and Carrie Miceli, PhD, UCLA (download presentation), shared a variety of potential avenues for circumventing the neutralizing anti-body issue. Of the following, a combination of plasmapheresis and immune suppression are the most readily available.
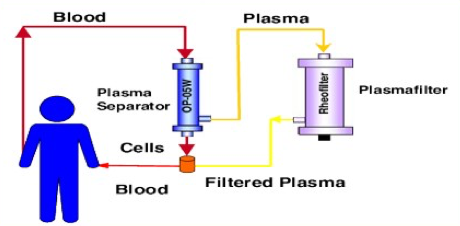
- Plasmapheresis – Similar procedure to donating plasma, blood is drawn and the plasma is separated. The plasma is then ‘cleaned’ of the anti-body and delivered back to the patient. Multiple rounds of plasmapheresis may be necessary to get below an anti-body titer threshold, but this is a fairly routine procedure. There has been pre-clinical data showing its effectiveness at reducing AAV titers for delivery.
- Immune Suppression – Using FDA approved immune suppression drugs in the lead-up to treatment that would help dampen an immune response and induce tolerance to AAV and dystrophin. PPMD is currently funding preclinical research investigating a number of these drugs and their impact on dystrophin restoration therapies. Other strategies are being pursued in other diseases and might be applicable to Duchenne.
- Designer AAV – An individual may only have antibodies to a single or few AAV serotypes (e.g., AAV5 or AAV9) so utilizing a different serotype of AAV may avoid an immune response. Directed evolution of AAV capsids could generate novel serotypes of AAV that the body has not been exposed to. Cross-reactivity of antibodies may hamper this approach (e.g., Anti-body to AAV5 may still target another AAV). Shielding of AAV involves using various chemical modifications to protect or hide AAV from the immune system during delivery.
Barry Byrne, MD, University of Florida (download presentation), discussed issues both around manufacture of virus and titer testing. Compared to currently approved gene therapies, such as Luxturna that is delivered to the eye, delivery to muscle requires an almost 10,000 fold increase in AAV. With this increase in viral load comes significant cost in manufacturing. Standardization of viral production and quality control will be paramount as these potential therapies in Duchenne move closer to market. There is also a need for comparison of the titer testing protocols between sponsors. Currently titer testing from one protocol is not applicable to another protocol, this includes any private tests performed for AAV titers. Standardization of these testing methodologies is necessary to help families plan for care and trial opportunities. The National Institute of Standards and Technology (NIST) is currently developing standards that could be used as a comparison point for industry.
The Patient Voice
PPMD’s SVP of Community Engagement, Ryan Fischer, presented preliminary data from the patient preference study. The findings of the study showed relatively high levels of risk tolerance that increases with disease progression. It also showed when making decisions on whether or not to join a gene therapy clinical trial, participants prioritized protection of muscle function over all other factors in decision making weighed against the progressive nature of Duchenne. Later this year you can expect a published report based on the findings of the patient preference study. This type of patient data can inform industry trial design, FDA decision making, and payer determinations (medical insurance).

A Patient Panel comprised of parents and an adult with Duchenne discussed what it is like to be restricted from current gene therapy trials. Each member had a unique experience, but shared similar wants:
- broader inclusion criteria,
- development of methods to treat those with neutralizing antibodies,
- expansion to non-ambulatory patients (Solid Biosciences has included non-ambulatory patients in their IGNITE trial)
We’d like to thank Colin Rensch, Colleen Labbadia, Jeffrey Bigelow, and Alison Hoke for their willingness to share their personal stories and shine light on issues facing families around gene therapy.
The Future of Gene Therapy
Tim Franson, MD, Chief Medical Officer, YourEncore, closed the meeting with a summary of the talks and expanded insights into needs for the field. The development of testing standards and validation of surrogate endpoints are key issues to the success of these gene therapy trials. Sharing of pre-competitive data between researchers and industry is important to the continued evolution of gene therapy as a potential therapeutic. A push for combination therapy is needed, as gene therapy will alter the disease progression of Duchenne, but not cure it. Finally, these are very exciting times as these technologies continue to develop, but they are still early in development, so although there stands to be great benefit, the highest priority lies within safety of the therapy. There also remains work to be done around potential avenues for circumventing the neutralizing anti-body issue and potential for redosing in the future.
Next Steps
This Gene Therapy Policy Forum provided an excellent dive into the current landscape of gene therapy. Bringing together experts in the field will help inform PPMD’s next steps in aiding the development of these therapies and finding avenues for treatment of all those with Duchenne. PPMD will continue to build on the learnings of this forum through a series of gene therapy webinars aimed at further educating the community later this year.


